Preprocessing
import os
from pensa.preprocessing import load_selection, \
extract_coordinates, extract_coordinates_combined, \
extract_aligned_coordinates, extract_combined_grid
To work with the biomolecule’s coordinates, it is often easier to first extract them from the simulation, i.e., remove the solvent, lipids etc. If you would like to calculate water or ion features, you need to calculate the corresponding density. This kind of preprocessing steps can be cumbersome but you usually only do it once and can then play with your data.
Based on MDAnalysis, PENSA’s preprocessing functions can handle many common formats of molecular simulation trajectories. You can start by using the scripts provided in the PENSA repository. Once you know how PENSA works, you can write your own scripts.
Files and Directories
In the following, we define the necessary files. For each simulation, we need a reference file (.psf for AMBER), a PDB file, and the trajetory.
To run this tutorial on another system, you’ll have to adapt the file
paths and names and, in case you need them, the
residue selections in the folder selections. We explain how they
work further below. Note that for some PENSA functions it is sufficient
that the derived features are the same while for others (especially
those that involve trajectory manipulation), all atoms need to be the
same. In our particular example, we exclude hydrogen atoms because
residue Asp114 is protonated in the BU72 simulation but not in the apo
simulation.
root_dir = './mor-data'
# Simulation A
ref_file_a = root_dir+'/11427_dyn_151.psf'
pdb_file_a = root_dir+'/11426_dyn_151.pdb'
trj_file_a = [root_dir+'/11423_trj_151.xtc',
root_dir+'/11424_trj_151.xtc',
root_dir+'/11425_trj_151.xtc']
# Simulation B
ref_file_b = root_dir+'/11580_dyn_169.psf'
pdb_file_b = root_dir+'/11579_dyn_169.pdb'
trj_file_b = [root_dir+'/11576_trj_169.xtc',
root_dir+'/11577_trj_169.xtc',
root_dir+'/11578_trj_169.xtc']
# Base for the selection string for each simulation
sel_base_a = "(not name H*) and protein"
sel_base_b = "(not name H*) and protein"
# Names of the output files
out_name_a = "traj/condition-a"
out_name_b = "traj/condition-b"
out_name_combined="traj/combined"
For this tutorial, we will save the processed trajectories in the
subfolder traj. We also create subfolders for other results that we
will generate.
for subdir in ['traj', 'features', 'plots', 'vispdb', 'pca', 'clusters', 'results']:
if not os.path.exists(subdir):
os.makedirs(subdir)
Coordinates
We have to ensure that from both simulations, we use the exact same
parts of the receptor for the analysis. Often, this will be easy and you
just provide a simple selection string for the corresponding segment.
For more complicated cases, we can use the function load_selection()
to generate a complete residue list from a plain text file. This file
should provide in each line the first and the last residue to be
considered for a part of the protein.
In the first case, we will extract all protein residues, assuming (correctly) that the same ones are present in both simulations.
# Extract the coordinates of the receptor from the trajectory
extract_coordinates(ref_file_a, pdb_file_a, trj_file_a, out_name_a+"_receptor", sel_base_a)
extract_coordinates(ref_file_b, pdb_file_b, trj_file_b, out_name_b+"_receptor", sel_base_b)
In some cases, you may have only one trajectory while in others, you may have several runs of the same simulation that you want to combine to one structural ensemble. This is why the trajectory argument can be either a single string
extract_coordinates(
'system.psf', 'system.pdb', 'run1.nc',
'receptor', 'protein', start_frame=1000
)
… or a list of strings.
extract_coordinates(
'system.psf', 'system.pdb', ['run1.nc','run2.nc','run3.nc'],
'receptor', 'protein', start_frame=1000
)
With the option start_frame, you can exclude the equilibration phase
already at this stage. Be aware that in combined simulations, there is
no straightforward way to exclude it later as it would require
bookkeeping about how long each simulation was etc.
Selecting Subsets of Coordinates
For some analysis types, we only want to use the part of the receptor that is inside the membrane. In this way, very flexible loops outside the membrane cannot distort the analysis result. We can manually construct a selection string in MDAnalysis format. Here, we use selections based on the definitions of transmembrane helices in the GPCRdb.
# Residue numbers (same in both simulations)
resnums = "76:98 105:133 138:173 182:208 226:264 270:308 315:354"
# Generate the selection strings
sel_string_a = "protein and resnum "+resnums
print('Selection A:\n', sel_string_a, '\n')
sel_string_b = "protein and resnum "+resnums
print('Selection B:\n', sel_string_b, '\n')
# Extract the coordinates of the transmembrane region from the trajectory
extract_coordinates(ref_file_a, pdb_file_a, trj_file_a, out_name_a+"_tm", sel_string_a)
extract_coordinates(ref_file_b, pdb_file_b, trj_file_b, out_name_b+"_tm", sel_string_b)
Loading from Multiple Simulations
If you want to combine data from different simulation conditions, you
can use the _combined version of the extraction function:
extract_coordinates_combined(). It takes lists as arguments for the
topology files, too. To use the same selection, “multiply” a list of one
string, as demonstrated below. For this to work, the two selections need
to have the exactly same atoms, so we mak a new selection below removing the additional hydrogen in simulation B.
# # Residue numbers (same in both simulations)
resnums = "76:98 105:133 138:173 182:208 226:264 270:308 315:354"
# # Generate the selection strings
sel_string_a = "not name HD2 and protein and resnum "+resnums
print('Selection A:\n', sel_string_a, '\n')
sel_string_b = "not name HD2 and protein and resnum "+resnums
print('Selection B:\n', sel_string_b, '\n')
all_refs = [ref_file_a]*3 + [ref_file_b]*3
all_trjs = trj_file_a + trj_file_b
all_sels = [sel_string_a]*3 + [sel_string_b]*3
extract_coordinates_combined(
all_refs, all_trjs, all_sels,
'traj/combined_tm.xtc',
start_frame=400
)
Densities
To work with the protein densities, we need to follow the standard density generation procedures for the input trajectory. Namely, centering on the protein of interest, making all molecules whole, and mapping the solvent molecules to be closest to the solute. To visualize the density featurization, the trajectories must be fit onto a reference structure. Note that the density featurization performs best for protein systems that are relatively rigid with sites that are spatially static, for example internal water cavities in membrane proteins. Here we demonstrate the preprocessing for water density, however the same procedure would be used for ions.
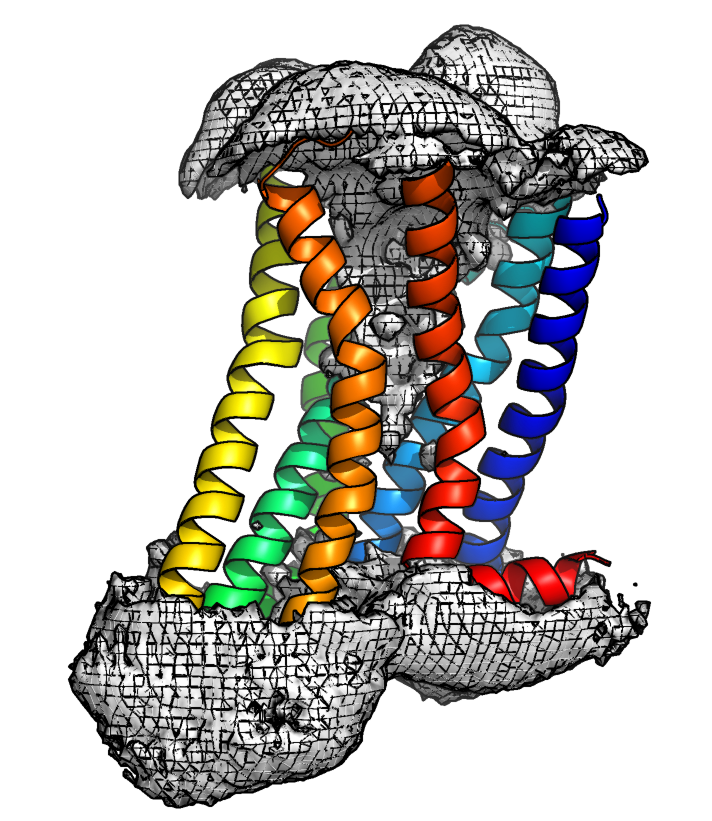
Files and Directories
We use the input files as defined above, and furthermore, we define a selection including the water residue name for the density. To featurize the water density, we must use a trajectory that includes hydrogens, however the density itself does not need hydrogens. It can therefore be useful to preprocess a trajectory including the entire solvent for featurization, and generate the individual densities from a smaller selection.
from pensa.preprocessing import *
# Base for the selection string for protein and all waters (OH2)
sel_base_water = "protein or byres name OH2"
# Names of the output files
out_name_water_a = "traj/condition-a_water"
out_name_water_b = "traj/condition-b_water"
Aligning Coordinates
As waters are not “attached” to the protein, water sites are defined spatially. Therefore to locate the same sites for comparative analysis across both protein ensembles, we have to ensure that the protein is aligned across both simulations.
We first extract the coordinates of the receptor from the trajectory.
extract_coordinates(
ref_file_a, pdb_file_a, trj_file_a,
out_name_water_a, sel_base_water
)
extract_coordinates(
ref_file_b, pdb_file_b, trj_file_b,
out_name_water_b, sel_base_water
)
Then we align the coordinates of the ensemble a to the average of ensemble b.
extract_aligned_coordinates(
out_name_water_a+".gro", out_name_water_a+".xtc",
out_name_water_b+".gro", out_name_water_b+".xtc",
xtc_aligned = out_name_water_a+"_aligned.xtc",
pdb_outname = out_name_water_b+"_average.pdb"
)
Extracting the Density
The density is then extracted from the combined ensemble, in which the solvent cavities are aligned.
We have the option to write out a pseudo-trajectory coordinate array to a memmap. This helps us avoid memory errors with large python arrays.
extract_combined_grid(
out_name_water_a+".gro", out_name_water_a+"_aligned.xtc",
out_name_water_b+".gro", out_name_water_b+".xtc",
atomgroup="OH2", write_grid_as="TIP3P",
out_name="ab_grid_",
use_memmap=True, memmap='traj/combined.mymemmap'
)
This density can now be used to locate and featurize the same water pockets in both individual simulations, even if a water site only exists in one simulation.